In a week’s time on June 19 the global health community will come together to discuss one of the least understood identified blood disorders - Sickle Cell Disease (SCD). This blood disorder has not received the high priority it needs including Sri Lanka largely due to the comparatively very small number of SCD patients we have. And despite the availability of diagnostic and screening facilities in most government hospitals, we still have several Sickle Cell pockets in the country where the disease is still widespread.
The Sunday Observer spoke to Consultant Haematologist and Head of the Haematology Unit, National Hospitalof Sri Lanka (NHS) Dr. Bhaddika Jayaratne to get more insights into the disease and how it can be treated, and managed with the proper medications taken regularly by the patients to whom it has been prescribed.
Excerpts
Q: While Sickle Cell Disease (SCD) is considered as one of the world’s foremost genetic diseases, many persons, don’t know what the disease is while even those with this condition remain unaware they have it. What is SCD? How is it transmitted?
 A. Sickle cell disease is a group of inherited genetic red blood cell disorders.
A. Sickle cell disease is a group of inherited genetic red blood cell disorders.
Red cells of these people who have sickle cell disease are rigid and deformed into sickle shape due to the presence of a mutated hemoglobin gene ‘HbS’. They are abnormal and called sickle cells. They lack the capacity to bind oxygen and break down prematurely. They also tend to clog blood flow particularly in small blood vessels. As a consequence, sickle cells generate multiple clinical complications of varying degrees of severity. In contrast healthy red cells are biconcave in shape and circulate easily through minute blood vessels carrying oxygen bound hemoglobin from lungs to peripheral tissues.
Q: Who are those most vulnerable to getting it- age wise and gender wise?
A. Sickle cell disease is an autosomal recessive inheritance in which both males and females are affected from birth. The severity of the disease and their manifestations mainly depend on the severity of the gene and the percentage of fetal hemoglobin in blood.
Q: Traits that help recognise Sickle cell disease?
A. Those who inherit two sickle genes from each parent get ‘HbSS’ or the homozygous sickle cell disease (SCD) which is the most severe form. Those who inherit one sickle gene from one parent and a normal ‘HbA’ gene from the other parent get ‘HbSA’ or sickle cell trait (SCT). People with SCT do not manifest symptoms and live a normal life. But they transform SCT to their children. Those who inherit the ‘HbS’ gene from one parent and ‘HbC’ from the other parent get ‘HbSC’ disease. This is usually a milder form of sickle cell disease. Those who inherit ‘HbS’ gene and Beta thalassemia from the other parent get ‘HbS-Beta thal’ (SBT or sickle beta thal). The severity of this form depends on the severity of its Beta thalassemia gene.
Q: What are the chances of someone with HbS who marries a person with similar traits to have a normal baby?
A. If both parents carry one copy of HbS gene, the chances for an unaffected or affected pregnancy is 25 percent. There is a 50 percent chance of an asymptomatic carrier state in this kind of an example.
Q: What are the adverse health impacts on the human body from SCD?
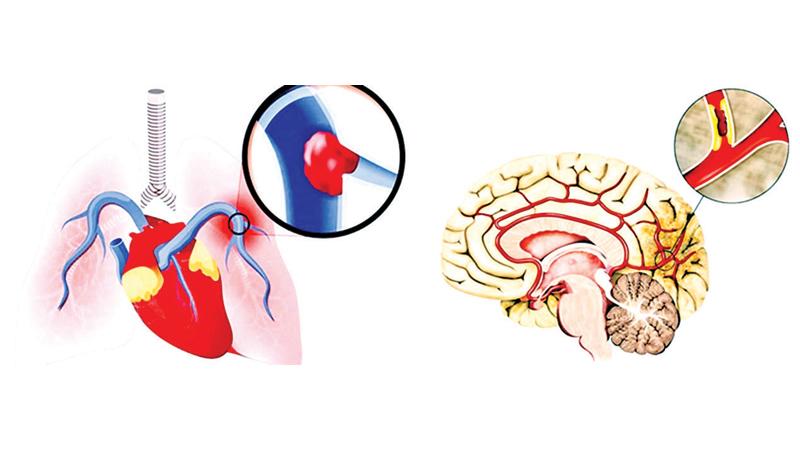
A. Patients can present with excessive breakdown of red cells in the vasculature that results in low hemoglobin level or anemia. They also could show yellow discoloration of eyes and urine. The complications could be severe and acute due to lack of supply of oxygen to tissues or obstruction of the blood flow particularly in small blood vessels. This results in damage to organs and episodic pain usually in the chest, abdomen, joints and bones. Persistent high blood pressure of the local vasculature in the lungs, a condition called pulmonary hypertension could lead to heart failure and even fatal outcome. Clogging of the blood flow in minute arteries of the brain can lead to stroke or transient ischemic attacks. Blindness could occur if small blood vessels to eyes are obstructed.
Chronic damage to liver, kidneys and pancreas can give rise to liver-renal failure and diabetes mellitus. Sickle patients also have a higher chance of experiencing deep vein thrombosis and pulmonary embolism which could result in death.
Q: Are SCD patients exposed to Covid-19 and other infectious diseases more vulnerable?
A. SCD is an immune compromised condition with high risk for respiratory tract infections. People with SCD infected by SARS-CoV2 virus have a higher risk of severe disease course and high case fatality rate. The acute vaso-occlusive crisis in SCD due to covid19 infection increases the probability of pulmonary embolism and acute chest syndrome. Anemia is an independent risk factor associated with the severe illness of covid19 in patients with SCD. All types of available Covid19 vaccines are safe for them. Among other common viral infections, Rhino virus infection is frequent in SCD patients. Parainfluenza 3 virus infection in them is reported as associated with adverse outcome requiring blood transfusions. H1N1 virus infection is associated with a milder form.
Q: Is it curable if detected at the onset?
 A. Allogenic Hemopoietic Stem Cell transplant (HSCT) from a matched sibling donor cures 85 percent of children with SCD if transplanted at less than 16 years of age.
A. Allogenic Hemopoietic Stem Cell transplant (HSCT) from a matched sibling donor cures 85 percent of children with SCD if transplanted at less than 16 years of age.
HSCT can be considered in children who have HLA matched donor, evidence of target organ damage and those who are placed on long term blood transfusions for any reason.
Q: What are the tests used to diagnose SCD and how are they done?
A. The Sickle hemoglobin can be confirmed by two methods of tests. The confirmation of the disease depends on the demonstration of HbS, HbF and HbA2 in blood. The commonly used test is hemoglobin capillary electrophoresis. Tests such as hemoglobin electrophoresis and high-performance liquid chromatography are used in only a few institutions in the country. The sickling test is a simple slide test used for the purpose of screening. These tests can be done in patients at any age.
Q: Are they available here as well? If so,where?
A. Yes, Diagnostic and screening tests are available at all teaching hospitals, some general hospitals where there are functioning hematology units and the Medical Research Institute. They are also available at private hospitals in Colombo and out stations.
Q: How do you keep the disease under control in patients with chronic SCD?
A. There is an oral drug called Hydroxyurea which is prescribed by hematologists and doctors in their teams to chronic SCD patients to reduce and prevent complications of sickle cell disease. It lowers the number of episodes of pain and acute chest syndrome by increasing total intracellular haemoglobin. It also makes the red cells flexible to circulate through the vasculature easily. This drug is safe to use on a long term basis and is available in institutions to prescribe under supervision.
We also offer medical advice to patients with sickle traits to avoid triggering factors such as, smoking, excessive use of alcohol and strenuous exercise and to be vigilant on maintaining hydration.. The prevention of infection and treating infections in patients with SCD is one of the key roles of the management plan, which includes recommended immunizations such as Hib, Pneumococcal and Meningococcal vaccines and Influenza vaccine.
Q: I understand you have a team of dedicated staff to do this? Who comprises this team?
A. At present there are about 78 members in the college of haematologists serving the country and keeping track of SCD patients as well as conducting special haematology clinics in the hospitals aiming at health education and improvement of the quality of lives of patients. These haematologists work together with medical laboratory technologists who cover the technical part of the diagnostic work up.
Q: Treatment wise—we are now moving forward into a high-tech era.. How far has new technology helped in improving survival rates in patients?
A. The Allogenic haemopoietic stem cell transplantation in SCD is the promise for cure as described earlier. Lentiglobin BB305 is a gene therapy drug that inserts functional human Beta globin genes to a patient’s stem cells and produces non-sickle hemoglobin. There are novel drugs such as Voxelotor that stabilises the oxygenated status of HbS and Crizanlizumab is a monoclonal antibody that prevents vaso-occlusive crisis. Exchange blood transfusions are done to increase the percentage of HbF up to a safe level in sickle cell patients prior to anesthesia and surgery.
Q: The Global Alliance of Sickle Cell Disease Organiations( GASCD0)) has said that this year’s themes for SCD are : “ Building and strengthening Global Sickle Cell Communities, Formalising New-Born Screening and Knowing your Sickle Cell Disease Status. “What is the Sri Lanka College of Hematologists (SLCH) doing towards achieving these goals?
A. The Sri Lanka College of Haematologists is the main stakeholder for establishing a register for haemoglobinopathies which includes essential data of sickle cell disease patients and carriers island wide under the patronage of the Ministry of health. This information regarding distribution patterns and clinical description of HbS gene in Sri Lanka will be helpful in understanding SCD better to initiate necessary health programs for patients in the future.
Q: Do you have Screening programs for new born babies?
A. If one parent is SCT, the other parent is screened at the time and if negative, the baby will be tested after two years of age.
Q: Does the community have a role to play in raising more awareness of SCD among people living in remote areas to whom the message many not have been conveyed?
A. Community support should be a mandatory part of the lives of people who suffer from any lifelong inherited disorder. They may need help, understand emotional reassurance, love and care at different stages and times of their course of the illness. SCD is one of the least known and identified diseases among Sri Lankans despite the fact that we still have several pockets of sickle cell disease in Sri Lanka e.g. Batticaloa, Puttalam, Mannar and Kurunegala. The Hambanthota districts which have reported the highest incidence of HbS gene. Organising education programs to disseminate information to all citizens up to the grassroot level through supporters, professionals, engaging community leaders and creating media strategy particularly for those who live in these districts would be constructively fruitful.
Q: Do you have a message for those striving to improve the quality of life of all our Sickle Cell Lankan patients ?
A. SCD is a lifelong genetic disease. It is hard for sickle cell disease patients to have a normal day to day life like other people. A long-term plan and comprehensive approach to improve outcomes and quality of lives of sickle cell patients is as important as pain and acute management of the disease and is the need of the hour.